En el No 28 de Vitae (Academia Biomedica Digital) de la UCV, mostramos siete casos de un tumor muy agresivo y poco conocido: “el Sarcoma Epiteliode”; esto sucedió en el año 2006, cuando ya el país estaba entubándose por “el socialismo del siglo XXI” y las posibilidades de crecer científicamente para los anatomopatólogos ya habían comenzado a perderse, y el rumbo de nuestra especialidad médica parecía destinado a torcerse, triste y lamenteblemente…
El Sarcoma Epitelioide inicialmente fue un tumor maligno descrito por Enzinger en 1970 y se presentaba como un tumor en el tejido celular subcutáneo o más profundamente en las manos o los brazos de adultos jóvenes con una alta recurrencia y con propensión a dar metástasis… ¿Por qué estoy aquí en este blog leído por un público heterogéneo hablando de este terrible tumor? Es buena la pregunta… Cada quien hallara su respuesta.
En aquel entonces (año 2006), revisamos nuestros archivos, los del Laboratorio de Patología Molecular NOVAPATH, entre los años 1998 al 2006), y detectamos siete ( 7 ) casos de tumores que después de su evaluación inmunohistoquímica habíamos diagnosticado como Sarcoma Epitelioide.
CASO No 1: mujer de 26 años con una lesión en la mano y la axila izquierda. La apariencia histológica sugirió el diagnóstico de Schwannoma. CASO No 2: una señora de 78 años con un nódulo en la región parotídea, considerada histológicamente como un carcinoma indiferenciado de la parótida. CASO No 3: un hombre de 36 años con un tumor en la región perineal interpretado como un Hemangioendotelioma Epitelioide. CASO No 4: paciente masculino con lesión multinodular en el tejido subcutáneo de la región glútea y se interpretó como un tumor vascular.
CASO No 5: una paciente muy joven con un tumor maligno epitelioide interpretado inicialmente como un melanoma. CASO No 6: paciente de sexo femenino con una tumoración en el dorso de la muñeca izquierda, operada con la impresión de lesión neural en el tunel carpiano, posteriormente interpretada como un Sarcoma Sinovial. CASO No7: mujer de 49 años con tumor sunmaxilar y en la revisión de la biopsia previa (No 6707-01) con el No. 1051-2001 en diciembre del año 2001 y presencia de inmunomarcaje para Desmina, Myo-D, Actina muscular específica y Vimentina apoyó el diagnóstico de Rabdomioma.
Ejemplos de H&E, EMA y Vimentina.


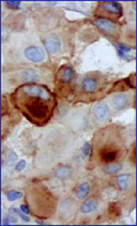
El propósito de este breve trabajo es demostrar los beneficios diagnósticos de una técnica que desde los años 80 revolucionó el diagnóstico anatomopatológico en el mundo y que comenzamos a aplicarlo en el IAP de la UCV en aquella década, pero su recuerdo hoy día sirve para destacar la importancia de la inmunohistoquimica en el diagnóstico de los tumores poco diferenciados.
En el caso especial de esta neoplasia muy maligna, el diagnostico nuestro estaba basado en las observaciones hechas sobre la transfección de las cadherina N y E en las células fusiformes de sarcomas murinos donde ese fenómeno les confería una apariencia epitelioide. En aquellos días se sabía igualmente sobre el efecto que provocaban la presencia de cadherinas en los tumores malignos de los nervios periféricos de aspecto epitelioide, en los nevus melanocíticos, y hasta en los linfomas anaplásicos cuando adquieren el fenotipo epitelioide. (ver referencias 1 y 2 ).
Existe una alteración genética que parecía ser específica del tumor rabdoide maligno (TRM), una alteración en el gen hSNF5/INI1 (SMARCB1) ubicado en el brazo largo del cromosoma 22. También se ha demostrado en el SE de tipo proximal, hallazgo que nos lleva de nuevo a relacionar como de un origen común a estos dos tumores con fenotipo epitelioide y rabdoide. El TRM es una neoplasia infantil de crecimiento rápido y poco frecuente que se produce en lactantes y niños pequeños y la cual por lo general, comienza en los riñones pero puede aparecer en cualquier parte de los tejidos blandos del cuerpo (en el cerebro se llama tumor teratoideo/rabdoide atípico) y con frecuencia se disemina precozmente.
La presencia de células rabdoides en algunos tumores malignos de partes blandas como el sarcoma sinovial, el leiomiosarcoma y el condrosarcoma mixoide extraesquelético, se ha asociado con un peor pronóstico en la evolución de estas neoplasias y quizá uno de los diagnósticos diferenciales importantes, es cuando el SE muestra células rabdoides que recuerdan al TRM, una neoplasia que, en la infancia, se ve en el cuello y en regiones paraespinales. El sarcoma epitelioide debe diferenciarse también de los carcinomas indiferenciados. En realidad, se sabe que las células rabdoides se pueden ver en diversos tumores epiteliales, como en adenocarcinomas del estómago, del pulmón y de los riñones.
También se ha demostrado la expresión de dysadherina en el SE; la dysadherina es una glicoproteína de membrana que actúa frenando la acción de la E-cadherina y se cree puede promover las metástasis. Examinada en 72 casos de SE se demostró un aumento de la expresión de dysadherina en el 71% de los SE proximales y en el 36% de los SE distales. El hallazgo de la expresión de esta glicoproteína sugiere un incremento en la motilidad y supuestamente en la agresividad de las células de SE, por lo que esto implicaría un peor pronóstico, aunque hay indicios de que el comportamiento biológico del SE, parece depender más de la regulación del ciclo celular a través del gen hSNF5/INI1 que de la expresión de sus moléculas de superficie.
Cincuenta y tres años después de que Enzinger publicara su artículo sobre el SE (1970) y que más tarde cuando en 1985 expandiera sus observaciones, el Sarcomo Epiteliode sigue siendo una entidad clínica y patológica específica, de curso indolente, y muy agresiva sin que haya sido posible hasta ahora determinar su histogénesis "El sarcoma epitelioide representa menos del uno por ciento de todos los sarcomas de tejidos blandos", dijo Richard Pazdur, M.D., Centro de Excelencia Oncológica de la FDA. “Hasta hoy, no habían opciones de tratamiento específicas para pacientes con sarcoma epitelioide y Tazemetostat proporciona una opción donde los beneficios del medicamento superan los riesgos ".
La extirpación quirúrgica es el tratamiento principal cuando el cáncer se localiza en un área del cuerpo. También se puede administrar quimioterapia o radiación. El Tazemetostat bloquea la actividad de la metiltransferasa EZH2 y puede ayudar a evitar que crezcan las células cancerosas. La enfermedad metastásica se considera potencialmente mortal para el paciente. Los efectos secundarios más comunes para los pacientes que tomaron tazemetostat fueron dolor, fatiga, náuseas, disminución del apetito, vómitos y estreñimiento. Se sabe que los pacientes tratados con tazemetostat tienen un mayor riesgo de desarrollar neoplasias malignas secundarias que incluyen: linfoma linfoblástico de células T, síndrome mielodisplásico y leucemia mieloide aguda.
Referencias: 1-Ashton-Key M y col. Cadherines in reactive lymph nodes and lymphomas: high expression in anaplastic large cell lymphomas. Histopathology 1996, 28:55-59.
2-Smith MEF y col. Epithelioid sarcoma: presence of vascular-endothelial cadherin and lack of epithelial cadherin. Histopathology 1998, 33:425-431.
Maracaibo, jueves 20 de julio del año 2023
No hay comentarios:
Publicar un comentario